能源化学理论研究及其应用前景浅述
知乎@咸球
原刊于 化术-知乎专栏
摘要 :能源物理化学和能源材料化学是能源化学中基础性、原理性和前沿性的研究领域,理论化学在能源材料的研究领域具有潜在的应用价值。本文分析了能源化学中的表界面问题以及能源化学过程中的重要理论等关键问题,结合能源材料化学中的重要理论研究问题,考虑我国煤炭为主,新能源逐步发展壮大的能源总体环境,提出了要逐步引导技术储备的产生和人才力量的支持,发挥日趋重要的理论研究在产业发展中的重要作用,并适时整合相关企业和研究机构在市场需求和技术储备的发展,保障产业和研究的对接,促进研究和产业发展的良性互动的发展展望。
引言
能源化学是能源学科和化学、化工甚至经济学、管理学等多学科交叉形成的一门新兴的关键科学。能源化学的研究主要目的在于提高能源利用效率和实现能源结构多元化,进而解决能源问题。中国科学院和国家自然科学基金委编著的《中国学科发展战略·能源化学》[^1] 将能源化学领域依据能源资源的不同和对能源体系和过程的支撑作用分为碳基能源化学、电能能源化学、太阳能能源化学、热能能源化学及能源物理化学、能源材料化学以及能源化学系统工程等多个三级学科。
其中能源物理化学和能源材料化学是能源化学中基础性、原理性和前沿性的研究领域。能源物理化学和能源材料化学的研究自然是实现能源系统工程中许多技术性要求的重点,也是各类能源形式的基础原理所在。能源问题的解决仰赖于能源体系(包括能源系统工程和能源器件)的高效构建,其中包含的复杂的能源过程同样是取决于能源材料的开发、制备以及其功效的合理发挥。
理论化学在能源材料的研究领域具有潜在的应用价值,在2011年出版,中国科学院和国家自然科学基金委所编的《未来10年中国学科发展战略·化学》[^2]中,业已指出了能源和资源的清洁转化和高效利用应当将理论、实验和计算相结合,通过分子调控和优化系统集成形成新一代资源高效转化和环境友好的新理论、新方法、新技术。该书业已指出了“功能导向设计、合成、理论和方法”、“化学反应动态学理论与实验技术”、“面向复杂体系、以从结构到性能预测为导向的计算方法的发展与应用”等一系列涉及如催化机制、界面作用、材料结构性质计算预测等能源化学和能源材料领域关键问题的重点研究方向。我国在这一领域的研究成果尚属起步[^1],本文结合我国研究现状和实际需求,整理归纳了能源化学理论研究相关领域比较前沿的研究内容。
能源物理化学的关键问题
能源物理化学是能源化学的重要组成部分,构成了整个能源科学领域所有应用、规划设计和前沿研究的基础。能源物理化学着眼于能源过程中的物理化学变化,针对能源科学关心的诸多理论问题进行研究和理论发展。
由于涉及很多界面反应,因此在能源物理化学中,表界面问题是相当重要的研究方向之一。同时,能源材料本身的功能性结构、纳米结构能源材料、半导体材料、复合纳米材料以及已经广泛实用的传统储能材料在能源化学过程中的过程理论问题还将是能源物理化学领域的重要理论课题。
能源物理化学中的表界面问题和相关研究
在物理化学中,涉及表界面的重要概念和理论有:表面张力、毛细现象、吉布斯界面模型、表面偶极、界面双电层理论、空间电荷层理论、表面润湿、范德瓦尔斯理论等。[^1]表面化学相关研究领域业已有近十位获得诺贝尔奖的科学家。
这些基础研究对于理解能源物理化学过程具有重要意义,并且随着能源科学和相关学科的发展,所涉领域的研究也呈现着新的发展趋势和特点。
经典的表界面研究和其局限
经典的界面模型一般讨论两“相”的共存边界,表面相是体系性质连续变化的一个过渡区域。同时,经典的相界面模型有吉布斯(Gibbs)相界面和古根海姆(Guggenheim)相界面模型两种(如图 2.1.1),这两种模型都是为了简化对于界面处物质不均匀分布的处理。
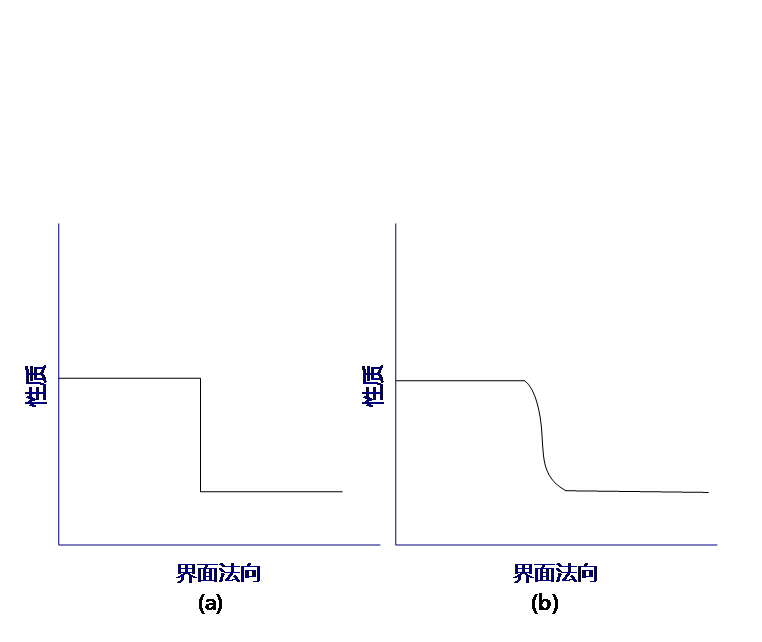
图2.1.1 吉布斯(Gibbs)相界面模型(a)和古根海姆(Guggenheim)相界面模型(b):吉布斯相界面模型中没有“相界面层”,物质性质变化是突跃的;而古根海姆相界面模型中存在一个过渡区域。
同时,表面化学中的三大公式[^3],即用于研究毛细现象的Young-Laplace方程式(2.1.1)、讨论表面曲率和蒸汽压关系的Kelvin公式(2.1.2)和讨论二元体系表面张力随组分浓度变化的Gibbs公式(2.1.3):
$$
\Delta p=\gamma \left( \frac{1}{R_1}+\frac{1}{R_2} \right) \qquad \left( 2.1.1 \right) \
RT=\ln \frac{P}{P_0}=\frac{2V_{\gamma}}{r}=\frac{2M\gamma}{\rho \cdot r}\qquad \left( 2.1.2 \right) \
\begin{cases}
\varGamma _{2}^{\sigma}=-\left( \frac{\text{d}\gamma}{\text{d}\mu _2} \right) _T\qquad \
\mu _i=\mu _{i}^{0}+RT\ln a_i \
\varGamma _{2}^{\sigma}=-\frac{a_2}{RT}\cdot \frac{\text{d}\gamma}{\text{d}a_2}
\end{cases}
\qquad \left( 2.1.3 \right)
$$
其亦是建立于连续介质宏观体系的热力学描述,可以看到这里不包含任何分子层面理化性质的物理量,压力、接触角、温度、化学势、表面能等均是分子体系的统计量。
此外,在界面双电层理论方面,不论是Helmhollz平板双电层理论或是Gouy-Chapman扩散双电层理论,抑或是考虑了离子半径修正的Stern双电层模型,其对于表界面电化学性质的理论阐述均是基于统计热力学或宏观统计规律的延拓。这些理论在表面化学刚刚发展起来的一段时期,可以很好地解释表面物理化学的稳态过程(但也主要是稳态过程),不过对于学界理解表面科学和相关应用研究的科学发展,其均是相当重要的理论基础。[^3]
随着能源科学应用向着快速、大量和非稳态过程研究的发展,经典的理论已经不能满足解释精细化的、动态的、存在弛豫的、快速变化的很多前沿研究成果的需要。同时,经典的表面物理化学研究对于动态过程,特别是与如吸附、表面物种的迁移等的研究(更多基于简单的单分子和表面的相互作用)相关的理论和实验手段还有很大的需求和发展空间。
能源(材料)表界面的基本物理化学特性的研究
当下,能源科学中,对于表界面物理化学的前沿研究主要缘于对于新型能源材料的开发需求,如纳米晶的自组装到从分子到材料表界面构建的过程,用于针对性构建具有特定要求的表界面结构,这类研究的跨度远远超过经典理论的解释范畴。
微尺度表面的热力学和动力学性质研究
在能源材料表面上,存在着物质和能量交换,其原子层级的微尺度表面性质对于实际应用研究具有十分重要的意义。如Dongsheng Li等关于氧化铁纳米颗粒表面在溶液中的生长控制的取向因素的研究[^4],通过高分辨透射电镜指出了分子簇在纳米颗粒表面的取向附着和后续的相互作用与生长机制。Na Tian等亦通过方波电位进行形貌控制[^5]实现了高指数面和高电氧化活性的四面体、六面体铂纳米晶的合成,并讨论了其微观机制和催化应用效果。
事实上,对于不同的微尺度表面结构的热力学和动力学性质研究非常广泛,同时也有多尺度模拟和贴合实际应用条件的模拟研究,不过总的来说。这类研究的系统性仍有待进一步提高,这仰赖于计算能力、理论方法和研究技术的进一步发展。窃以为相关研究无论是在学术界还是工业界,都会是越来越成体系、成规模的,当然这也会有助于微尺度研究方法的更加成熟和相关仪器设备的工业化和规模化生产,形成良性循环。
环境介质如气体吸附、液体环境、异质结的影响和电子态的调变作用
为了实现对于具有特定要求的表界面的生成,需要研究从原子团簇性质、连续相物理化学性质到团簇和连续相界面相互作用、复合材料的实际反应性能表现以致于复合材料的反应动力学模拟等。目前由于理论计算能力的限制,研究中更多的会着眼于某一个关键过程的理论研究和对于全过程的各种因素的实验研究。
如Vojislav R. Stamenkovic等[^6]通过对比Pt3Ni(111)与Pt(111)表面的催化活性,利用表面敏感探针和DFT计算研究其在氧化还原反应中的结构敏感性、原子组成偏析分布和合金化对于表面化学性质的影响。他们的研究证实了表面电子结构性质特别是d-带态密度的结构敏感性。另外,J. A. Rodriguez等[^7]研究了Au(111)表面上生长的CeOx和TiOx纳米颗粒在水煤气变换反应(water-gas shift reaction, WGS)中的不同活性,并研究了其反应机制。Qiang Fu等研究了限制在纳米尺寸基质中的配位不饱和亚铁(coordinatively unsaturated ferrous, CUF)位点,并通过表面测量技术和DFT计算证实了其在非均相催化反应中,有氧原子出现的反应机制。
传统领域的物理化学研究主要是基于刚球模型和宏观周期性体系,这些理论无法满足解释纳米尺度和动态变化特征的能源化学过程的需要。[^1]同时,当前的能源科学研究中对于表界面问题的阐述正处于单一维度到多维度、静态到动态、模型体系到功能器件过渡的时期。[^1]当前对于理论计算模拟的需求进一步增加,同时也对于系统性的理论模型和研究技术提出了更高的要求,因而适应新的能源科学研究的理论和研究方法的开拓就成为了能源物理化学进展的关键问题,并将影响到未来能源科学应用的发展,具有举足轻重的地位。规模化和专业化的理论研究和成熟可靠的实验研究的良好结合,更是学术界和工业界共同的发展趋势。正如材料研究从试错、经验到精细化、理论引导技术研发的发展历程,能源材料的基础研究更应当尊重其面向社会生产实际的特点,建立面向需求的研究体系。
能源材料表界面结构调控修饰
能源材料与其他材料的一大不同,就在于其表面具有面向化学反应的纳米级功能化修饰组织,同时由于能源材料在实际,更多是有多个不同组件结构组合使用,材料更会和反应介质接触,产生新的物理化学特征,因而格外要考虑和环境介质的相互作用。
在材料表界面结构修饰中,尤为重要的是在固体表面上进行纳米结构的生长、表面的合金化和表面的纳米化等,如铝合金表面的纳米晶化与其生物相容性的研究[^8]、非晶态金属合金生成时的纳米晶化机制[^9]等。这类研究均指出了材料表面结构调控修饰的重要微观机制原理,在能源科学的研究中,具有重要的基础理论意义,不过显然,目前这类研究由于计算模拟能力的限制,更多地研究仍着眼于实验手法的尝试,本文不做进一步探讨。
适用于微纳尺度表界面体系和相关物理化学过程的新理论和新方法的发展
为了能够更充分地研究非平衡转化、表界面体系的多尺度方法和电子激发与动力学衰减过程等能源科学领域越发关心的问题[^1],在传统的理论研究和计算化学研究的基础上,发展包含时间、温度、压力等能源科学关心的环境参数的新的理论研究方法[^1]和简便易行有效的新模型也是未来的研究重点,这些问题实质上已经不仅限于材料和化学领域的范畴,还涉及复杂的化工工艺、实际工况的问题,特别是能源材料中的电子激发与动力学衰减过程问题,尽管研究艰难,但极有可能是未来研究的突破点。
能源化学过程中的理论问题
均相热化学反应机理的理论研究
研究范式
传统的反应机理研究中主要是以物理化学实验手段推测反应相关物种在反应过程中产生的中间体或过渡态等,并讨论电子效应、立体效应、取代基效应、溶剂化效应等对于反应的动力学、热力学、决速步骤、立体或区域选择性的影响。常采用动力学测定、中间体捕捉和同位素方法等,近年来又发展出了原位检测和实时光谱分析等方法。
通常情况下,基于理论和计算化学确立反应机理的正确过程如图 2.2.1所示。[^10]

图2.2.1 基于理论和计算化学确立反应机理的正确过程[^10]
正如《中国学科发展战略 理论与计算化学》[^10]书中指出的,传统的反应机理研究中,存在着无法给出多步反应的完整势能面、无法给出高活性反应中间体的结构信息、无法明确各种选择性与反应途径能量及过渡态结构间的关系、无法判断多种可能反应途径中能量合理的反应途径以及可能无法用实验解释无法实现或未实际发生的反应途径等问题。其实就是缺少对于反应体系的全面描述,或是研究的信息记录存在损失,并难以复用至其他研究中。这一范式还可参照各类反应数据库,继续细化研究得到的信息维度。
研究现状和相关进展
在反应机理研究中,尤为重要的是考虑所有可能反应路径中,各个势能面的误差控制,这对于计算化学的精度提出了更高的要求。由于在较大分子体系的模拟中,色散矫正具有重要的影响[^11],因而以此为代表的含色散矫正的DFT-D方法[^12]获得了广泛应用,未来应有更多的更精确的计算方法被发展出来用于复杂体系的研究,甚至是采用化学信息学中的统计手段解决误差控制的相关问题。
在溶剂模型的处理中,目前广泛使用的是极化连续介质模型(polarizable continuum model, PCM),其以溶剂的介电常数作为一个重要参数进行处理[^13],相应地也忽视了对于溶剂酸性、碱性、极化性等参数的考量,Claire S. Adjiman等[^14]通过多个参数的拟合,实现了对于溶剂反应的更准确地描述和反应的优化,应当说明,各种各样的统计手段正在快速发展以适应复杂体系动力学研究的庞大计算量和高精度要求,其前景尚不明朗,但机器学习等成熟的统计分析方法应当是一种实现精度和效率平衡的途径。
前景
对于能源化学,由于大量的化学反应的存在,如能从理论上理解包括均相催化、含能物种的变化、溶剂对于实际使用的影响的具体细节,对于能源科学的研究可以提供相应的理论依据,并可以进一步协助需要调控相关反应性质和解决副反应问题等的实际应用研究。
考虑到计算化学在反应机理的研究中业已取得相当的重要进展,如能实现对于应用尺度下能源化学问题的解决,并将理论与实验良好的结合,则将有力地推动化学理论、应用化学和化学工程等向依托规律设计和按需研究的方向发展。
多相催化理论研究进展
在能源科学中,有包括但不限于催化氢化、催化裂解、催化重整、水煤气生成、光敏化电池等的应用了多相催化技术,并对于催化剂的研发有着越发重视的需求。
结构-性质关系
一般认为在多相催化中,催化剂发生作用是依赖于其界面处的活性位,而这些活性位在理论上通过对于负载金属纳米粒子被近似为静态的多晶颗粒并含有晶面、边角、台阶和纽结等多种表面结构,并可以对这些具有特点的表面结构进行独立的研究。研究可以从实验研究和理论模拟两个角度认识纳米粒子的催化活性,不过在模型中,表面原子组成、排列方式、配位数等原子结构特征都会影响到特定催化反应的活性和选择性,而且对于不同反应机理,其影响有较大差别。
结构和性质关系的研究中,研究最广泛的是过渡金属,包括讨论费米能级处的态密度对于催化活性的影响、d带中心理论、几何结构敏感性反应、催化剂形貌等。同时通过理论计算,帮助了从原子尺度上了解多相催化中催化位点的电子性质、几何性质对于催化反应中分子活化过程的理解。
催化剂设计的理论依据
多相催化中的BEP关系(Brønsted-Evans-Polanyi relation)就是通过大量的DFT理论计算,分析一系列催化基元反应获得证实的[^15],其是一种热力学判据,表明物种的吸附能和反应活化能间存在着线性关系。同时BEP关系反映出表面反应过度态结构与反应终态非常接近,过渡态能量由反应终态能量决定的定量规律。[^16]
应用BEP关系,可以进行如合成氨的催化剂的筛选[^17]、乙烯加氢反应的催化剂筛选[^18]等,值得注意的是,在这些工作中,所进行的筛选工作是先于实验的预测,这正是未来的能源类催化材料所期待的一种研究模式。将DFT理论计算得到的数据和微观动力学模型良好的结合,可以使研究应用材料的工作更加具有系统性和实际意义。另外,基于此类数据分析工作的大数据平台和化学信息学也有一定进展,将在3.3节简介。
固液界面催化的理论方法
常见的固液界面会存在界面双电层,经典力学在处理固液界面时,采用点电荷和球形模型模拟溶液一侧[^19],采用硬球和软球模型模拟固体界面[^20];同时也有正则蒙特卡洛(canonical Monte Carlo)[^21]、Wertheim-Lovett-Mou-Buff(WLMB)积分方程[^22]、修正的泊松-玻尔兹曼方程(modified Poisson-Boltzmann)[^23]和分子动力学(molecular dynamics)[^24]等,都可以用于研究和描述固液界面的动态结构和双电层结构等。
光催化反应的理论研究
在能源科学中,光催化反应出现在光分解水制氢、二氧化碳转换等技术中,目前光催化反应主要使用金属配合物和以二氧化钛为主的半导体作为催化剂。
光催化过程中所涉及的材料激发态是Kohn-Sham方程不能描述的。另外,对于交换关联作用进行了近似的DFT有极大误差的部分,处理激发态等问题一般采用Hubbard参数U[^25]进行处理,不过这也存在争议。而在LDA交换能中引入Hartree-Fock交换成分构建的杂化泛函可以改善计算结果,不过结果也依赖于HF成分的多少。而格林函数的GW近似方法[^26]及其多体微扰理论可以较好地实现光催化过程中材料能级性质的计算,不过其往往存在非自恰和计算量过大的问题,限制了在比较大的体系上的应用。GW近似也有部分使用机器学习方法进行研究的探索,相关研究值得更深入的理论研究者关注,本文不做讨论。
在实际的光催化理论研究中,往往采用分子动力学和蒙特卡洛研究催化材料表面的结构和界面作用特征,然后以DFT计算复合体系的电子结构特征和几何特征,最后通过多体微扰理论格林函数方法得到体系的光学性质和光生载流子产生分布图象,可以较好地描述光子-电子、电子-电子和电子-声子相互作用。[^10]
煤转化过程的理论研究
在煤化工催化过程中,CO的高效转化和利用是核心,通常采用Fe、Co、Ru作为费托合成(Fischer-Tropsch synthesis, FTS)的主要催化剂。[^27]其中Fe基催化剂由于价廉易得tngshi 有较高的水煤气变换反应活性而成为了广泛使用的催化剂。
费托合成的机理
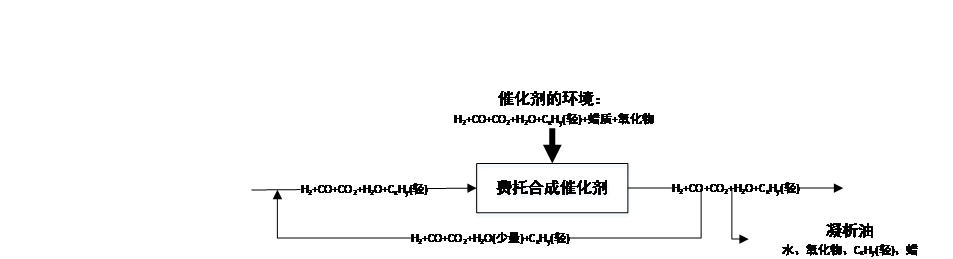
图 2.2.2 复杂费托合成示意图[^1]
第一种机理是表面碳化物机理[^28][^29][^30],即反应物CO与H2均是以解离吸附存在于催化剂表面,反应生成CH2进而结合或互相插入实现碳链延长、链终止获得烷烃或烯烃,不过这一机理不能解释含氧化合物的生成。同时有烯醇机理[^31],这一机理中CO是分子态吸附,并与解离吸附的H原子结合生成HCOH,进而聚合脱水或继续加氢脱水等,其以加氢链终止得到烷烃或烯烃,烯醇机理可以较好地解释反应中含氧产物的生成,但尚无确凿证据证实这这个机理的部分细节。此外还有CO插入机理[^32],是由CO插入到M-H键中形成醛,进而加氢得到CH3,由CO继续插入到M-C键中实现链增长,这一机理的存在需要更多证据的支持。一般认为在真实的反应中,以上机理应当同时存在,如在Fe5C2(001)表面,碳化物机理和插入机理是共存的[^33]。
煤转化的催化剂理论设计构想
对于煤转化反应有很多的催化剂理论设计研究工作,并且大都可以基于DFT进行计算[^34][^35][^36][^37]研究特定的催化剂和反应条件的实际效果,提出了很多具有实际意义的模型,成熟的反应机理研究模式已经建立,并可以解决应用中遇到的部分实际问题。不过需要指出的是,由于这类转化涉及到的反应体系和计算方法的限制,有很多的研究存在系统性缺陷,阻碍了实验-理论的对比参照,这也是DFT研究的传统问题。如想实现系统化的煤转化催化剂设计,基于现有的大量实验数据和理论计算结论,整合实现实验和理论研究的相互印证不仅具有理论价值,并且由于工业的一定需求,也具有其他研究不具备的经济可行性。另外,建立作用机理和理论计算的数据系统和数据库,形成标准化的计算和理论分析,将极大的帮助当前理论研究和催化剂设计筛选的有序研究,可参照3.3节的部分内容。
燃烧反应机理
燃烧过程作为能源科学的一个传统课题,从微观机理到宏观燃烧热流的研究对于包括航天航空工业在内的很多重要尖端工业技术领域有着重要意义,燃烧反应的机理作为燃烧理论的基础,相关研究可以视为一切研究的基石和未来研究的突破口,限于燃烧反应的复杂性和计算能力、模型适用性的限制和进来相关理论计算领域的进步,燃烧过程的理论研究有着新的潜力。
燃烧过程的基元反应研究
传统上,燃烧过程的模拟需要使用半经验计算的热力学和动力学数据,然后通过几类烃物种的替代燃料代替实际燃料,结合动力学实验数据验证反应类型和机理分级结构构建的替代燃料组分的详细机理。
燃烧模拟中所使用的热力学参数不仅是模拟的关键参数,同时关系到反应速率的准确性,即是通过热力学平衡计算得到逆反应动力学参数。常用的热力学数据库或手册有Ruscic提出的基于主动热化学表[^38](active thermochemical tables, ATcT)的数据库、CRC化学物理手册、NIST热力学数据库等。同时也有使用高精度从头计算方法构建小分子燃烧的精确的热力学数据库[^39]可以作为基准数据应用到机理研究中。
而动力学计算(主要是速率常数的计算)都依赖于势能面的计算,而势能面的计算中,对于精度,特别是涉及多参考态反应的计算是难点问题,而速率常数的计算还受到配分函数谐振子近似的误差影响,特别是在高温条件下非谐振效应的影响以及低温时隧道效应的影响。[^1]
在复杂燃烧反应的机理研究中,即替代燃料组分的详细机理过程这方面的研究中,通常希望计算机程序自动生成完整的基元反应和中间物种,如可用于550~2200K条件下的烷烃、烯烃、环烷烃、醚、醇和酯的EXGAS程序[^40]。
本文认为相关基元反应的基准数据在当今计算能力已经显著提高的大背景下,已经有使用更充分的、精确的理论手段进行计算的必要性,不过若是单纯的数据库建立可能在学术界暂时没有“研究价值”,或许在计算资源进一步发展后能够逐步建立扩展相关的基元反应数据集,结合人工智能的日益成熟,将助力更普遍的反应机理研究。
燃烧反应的分子模拟
由于燃烧反应模拟中所涉及到的分子体系的复杂性,量子化学计算尚不能得到完整且处处可靠的反应体系势能面,而能够快速有效处理较大体系的分子力场方法成为了燃烧反应分子模拟的重要方法。其中ReaxFF力场[^41][^42]通过计算由键长、键角、二面角、共轭、库伦、范德华和矫正项为参数的复杂函数得到原子间相互作用,可以实现周期表中一半以上的元素的通用反应力场参数,用于高温(3000K以上)条件下燃料在纳秒级时间内点火的模拟,尚不能实现真实条件(约1200K)下毫秒级时间尺度的模拟。随着计算技术和数据精度的进一步发展,燃烧反应的研究实现实时应用层级的模拟和实验-理论有序结合可能最具实用价值。
能源材料化学的理论研究
能源材料化学的研究主要涉及能源材料的合成、制备和其高效利用。《中国学科发展战略·能源化学》[^1]一书从新材料制备的角度,提出重点发展的研究方向包括功能介孔材料的制备、金属纳米结构的制备、二维半导体材料的制备、复合纳米结构的制备;本节关注材料结构和性质的关系,主要基于材料领域从理论模拟到指导实验的方法,综合叙述能源材料化学中的相关理论和原理的研究进展。
材料制备中的关键技术和原理
在材料制备中,特别是涉及到能源化学物理过程的催化表界面过程中,有大量基于结构和机理导向的纳米结构催化剂的设计。同时因为可再生能源的发展,大量基于新的原理的催化剂仍在不断地被设计出来。[^43]
在用于前沿催化过程的纳米界面设计工程领域,Zhi-cheng Zhang等[^44]总结出了金属-氧化物界面合成中的尺寸影响、支撑类型、分布、多金属连续层等的影响,以及金属核壳结构、异质结构催化剂,和金属-碳化物/硫化物/氮化物复合催化剂等的合成与应用,并分析了在纳米碳管等限制性空间架构中金属的催化作用和合成明确分界物质界面的方法。可以确信的是,在催化材料的研究中,不同材料和相的界面间的相互作用对于催化过程有着重要的影响。这种影响包括电子传递、异质结生成和活性位点等。
能源材料研究的理论研究
材料模拟的问题核心
《中国学科发展战略: 理论与计算化学》一书中,总结了材料科学对理论计算化学的挑战[^10],包括了复杂体系结构预测、材料功能定量且可靠的微观机制、材料生长的微观机理与动态演化三大问题。
复杂体系结构预测的核心在于多维势能面的准确分析,基于量子化学和统计力学,已经发展出了拓扑模拟、数据挖掘、随机取样、模拟退火、遗传算法等一系列高自由度体系分析的方法,其核心都在于加速取样以减少因为过多的单点能计算引起的耗时。而对于复合材料、界面体系、介孔材料和分子自组装体系,因为其非共价相互作用、缺少结构周期性的困难,尚不能应用结构搜寻方法减少预测时间,本文大胆预测这一方面算法优化的必要性可以从信息论的角度考虑进一步的研究。
而功能材料预测的微观理论领域,在材料对于力、热、光、电、磁等外场的响应中,材料电子结构的模拟,特别是激发态、散射/弛豫等的演化和响应正是材料性能预测的主要手段。具体到应用上,这方面有有机分子电致发光效率的提升、钙钛矿太阳能电池能量转换效率的提升和低热导高电导的热电材料的设计等。前文已经提到,这方面的基础计算研究仍面临精度和方法误差问题,因此大多数情况下还仰赖于实验的不断证实。
在材料微观生长机理和动态演化领域,由于模拟手段无法涵盖整个生长过程,因而基于第一性原理计算的模拟方法很难实现对于材料生长反应过程的完整预测,因而需要的是自洽的多尺度耦合理论方法,以合适、合理的模型研究材料生长过程,并获得完整的基于第一性原理的精准描述,进而结合数据挖掘和控制优化等方法实现对于生长方案的理论设计。当然,有效的数据挖掘和体系化的研究需要有全面精确、标准化的实验记录过程用作验证和进一步研究的基准,本文认为像Chemputer一类的标准化实验工具是潜在候选,但其适用范围目前来看还很小。
复合材料表界面的理论模拟
表界面分子及分子聚集体的电子结构
在表界面电子结构的计算中,主要使用簇模型(cluster model)和周期性表面模型(slab model)处理。如研究原子氢在簇模型石墨表面上的吸附[^45]使用了近似的晕苯(C24H12)作为替代石墨(0001)面的模型,类似的近似对于其他体系也适用,不过存在误差,并且因为忽略了除了簇原子以外的作用,会产生局部固体电子结构的定域化,无法获得准确的周期性性质。
而周期性表面模型可以用于如α-Al2O3的(0001)面的性质计算表征[^46]及Pd(111)表面上C、N、O原子的结合状态的研究和其上的水分解反应[^47]。不过此类模型没有充分考虑材料表面的缺陷、皱褶、相对滑移的存在有无影响,仍可能有进一步的研究空间。
此外,表界面处的电荷分布和极化作用的理论描述,对于如有机太阳能电池、(导电)高分子材料等的有机分子高度无序界面电子结构的描述也有进一步发展的需求。
表界面分子组装、多孔材料的模拟
自组装作为包括成膜材料、功能分子、胶体、陶瓷等在内的分子工程技术的一个重要构建基础,理论计算模拟可以使研究人员在构建自组装体系时减少盲目探索,更多地进行结构和性能关系的理解。而自组装体系中理论计算研究的难点在于精准描述分子间作用力并良好的处理溶剂的影响,并要综合考虑动力学稳定性的动态过程。对此,通常使用粗粒化模型进行简化,减少其作用中心以实现更广的采样计算,具体的方法模型有如MARTINI粗粒化力场方法[^48]、反向玻尔兹曼方法[^49]、多尺度粗粒化方法[^50]等。本文认为生物领域能够使用Foldit游戏助力蛋白质的研究,而高分子材料有很多类似性,或许也可以发挥相关但更有侧重的研究模拟思路。
催化材料
在催化材料化学性质研究中,通常着眼于在催化材料的活性中心处,底物分子和活性中心的电子结构和性质计算,目前广泛采用密度泛函理论研究相关的热力学和动力学性质。不过由于材料研究的复杂性以及计算能力的限制,在新的材料研究理论和计算方法的研究中仍有进一步发展的空间。[^10]
总体上,根据技术原理和应用范围的不同,材料研究的计算方法可以分为基于材料数据信息系统的预测、基于计算材料学理论的模拟方法两大块。前者是基于实验或模拟研究的大量数据生成的数据库,通过专家系统等人工智能技术进行数据挖掘等预测,其依赖于大量的数据积累。而后者在材料的不同尺度上又有着不同的计算方法。
在计算材料学中,根据研究对象的尺度大小,可以分为纳米尺度(纳观尺度)、微观至介观尺度、介观至宏观尺度等。其中在纳观尺度和微观尺度更注重对于材料化学性质的研究。如在纳观尺度上,常用第一性原理计算方法(包括Hartree-Fock自洽场方法和密度泛函理论方法),特别是密度泛函理论处理研究涉及过渡金属的材料电子结构和性质关系,但其对于计算能力的要求较高;而基于经验模型的分子动力学方法常用于处理至多数万至数亿个原子在纳秒量级上的动态变化过程。[^51]由于能源物理化学过程涉及更多的在微观到介观尺度的变化,因而结合经典力场、粗粒化模型和连续介电等模型的多尺度计算方法和计算谱学方法的发展,更有实际的重要性。[^1]
另外,在材料工程科学研究中,常见的理论计算方法包括Monte Carlo法、分子动力学方法、元胞自动机方法和有限元方法,主要研究着眼于材料在不同尺度下的力学、机械性能等。[^52]本节虽然着重讨论了能源材料的化学性质的研究方法,但如果要使能源材料理论研究能在实际工业应用中发挥作用,就需要更多地考虑材料工程科学方面的计算方法,充分考虑材料应用时的宏观实际物理和化学条件,特别是化工生产中涉及机械作用的工况。
理论计算指导材料制备和研发
按照《未来10年中国学科发展战略·材料科学》的设想[^53],材料科学按照学科特点可以分为材料设计与计算模拟、材料制备与加工、材料结构与相变、材料结构—性能关系和使役行为四个领域。材料计算与计算模拟指的是从20世纪90年代开始的,从相变设计、微力学设计、纳米设计、量子设计等不同层次的材料设计。而且在材料科学的四个领域中,计算模拟的手段都各有侧重,将共同助力材料研究的深入探索与研发。
在《未来10年中国学科发展战略·材料科学》[^53]中,指出了从“材料成分、结构与性能”、“制备加工过程与工艺设计计算模拟和设计”到“材料使役行为的计算模拟与寿命计算预计”的计算材料学研究发展态势,同时指出了我国发展计算材料学需要着重发展高效多层次高精度第一性原理计算方法、交叉功能材料的计算探索仿真和新型有机功能材料模拟和计算设计、材料成形加工的多尺度计算模拟仿真、复杂使役条件下材料损伤演化和寿命预测以及材料数据库与知识库等优先支持的研究方向。
需要指出,在世界范围内,使用计算机等信息科学工具进行材料研究已经是比较普遍或者说十分具有前景的,比如用于材料性能计算的分布式大数据分析架构pymatgen[^54]等。此外,本文预测,有可能用于进一步材料性质和理论化学研究的信息分析工具还可能包括深度学习框架TensorFlow或Pytorch等,不过其学习和研究成本还比较高。而能源领域至少在可控核聚变商用之前,都将有大量的资金投入,而信息分析产业在世界范围内已日趋成熟,或许将对能源化工产生一定的产业研究动力,甚至产生商业化的数据服务,不过由于信息服务业从业者的竞争优势客观存在,本文并不看好相关领域产业应用成果能很快出现。
中国的能源市场和研究领域
在2007年,中国占世界20%的人口,而消费了世界16.8%的能源,并创造了世界19.8%的GDP总量,而中国的目前人均能源消费和人均GDP仍低于世界平均水平,并处于高度依赖能源的发展阶段。中国同时也面临着来自于多方的的能源资源威胁论:一些国家对中国的国际能源活动横加阻拦,国内也有一些学者妄自菲薄,认为中国是一个能源极度浪费的国家,认为应当通过节衣缩食解决能源问题。好在已经有不少能源著作开始用科学的态度正视中国的能源问题,客观评估未来能源趋势。[^55]我国的能源结构主要呈现着结构依赖煤炭,新能源蓬勃发展,清洁能源份额逐步扩大等特点。
我国能源结构和市场情况
能源生产和消费情况
能源生产
2016年,我国能源生产总量为346000吨标准煤,其中原油和原煤比重继续下降,天然气、一次电力和其他能源比重继续上升。(见图 4.1.1)而根据国家统计局的数据,2017年中国能源消费总量为449000万吨标准煤。我国的能源供给仍然需要大量进口。
我国煤炭资源储量丰富,然而煤炭资源分布不均、地质赋存条件复杂、煤炭的科学合理的生产能力受到多种因素制约,《中国能源中长期(2030、2050)发展战略研究》指出[^56],考虑安全生产、水资源和生态保护等硬约束时,我国可能形成的安全高效的科学产能最多在29亿吨左右,考虑运输制约时将不到25亿吨,如果只考虑安全和机械化约束则可能达到35亿吨左右,其亦指出建议合理产能控制在30亿吨煤炭之内,远期以25亿吨为好。而对于我国可采石油资源量,比较有把握的估计为150亿吨,并还有油页岩油、油砂油等非常规石油资源。[^56]然而我国的石油产量难以大幅度提高,主流意见是以低于2.3亿吨为佳,并应略低于此值。由于我国的石油资源赋存条件较差,开发生产成本普遍偏高,因而在现有国际油价较高水平下我国的石油开发生产尚有充分的投资条件,然而若国际油价进一步回落,我国的石油产量和石油工业发展将受到较大挑战。我国的天然气资源和石油资源大致相当,而我国今后的天然气产能还可以继续提高,并且具有较好的价格竞争力。[^56]在水电、核电等领域,重要的制约因素是环境影响的验证研究和舆论价值判断,风电和太阳能发电领域的输电调度费用是成本中可以进一步降低的部分,充分的政策支持可以推动这些领域的发展。而在生物质能和其他可再生能源的开发供应中,由于受到多种条件制约,不确定性较大,一般应作为远期的可能替代能源进行研究储备。
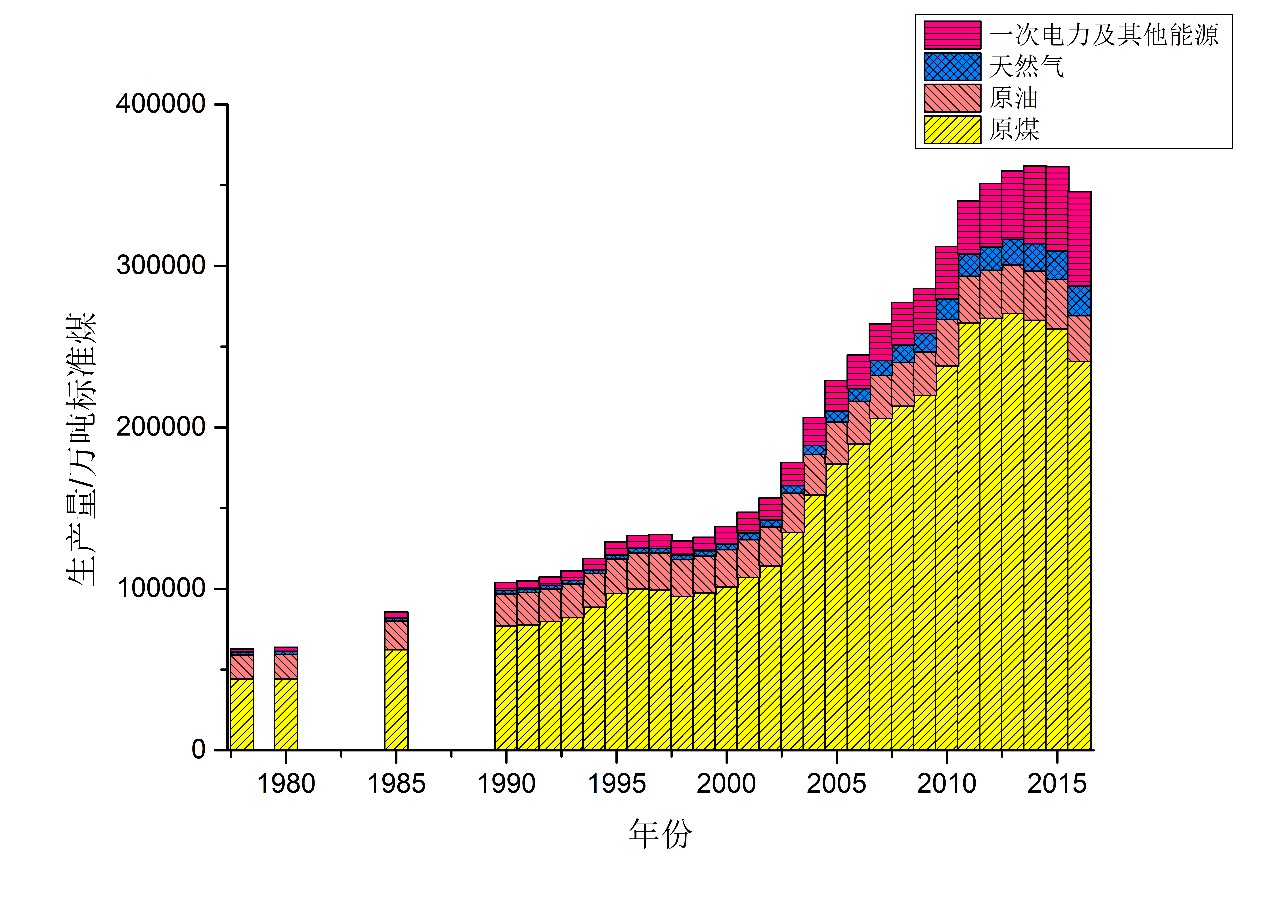
图 4.1.1 我国能源生产结构(截至2016年,数据来源:国家统计局《中国统计年鉴2017》)
在能源生产领域,由于生态环境的诉求,未来中国的能源结构将更多的朝着环保、清洁、低碳化发展,煤炭和石油消费占比将逐渐降低,但短期内煤炭作为中国主题能源的地位难以发生重大转变,因而要更多地发展煤炭情节开发利用技术,如煤直接液化、煤间接液化和清洁煤技术领域等。对于我国尤为重要的是技术落后、资金投入不足、政策支撑保证等的解决。[^57]其中技术领域除了基于现有生产条件的进一步优化改良,还有开发新的生产条件工艺,这就与能源物理化学的研究密切相关,并具有长远的社会和经济效益。
能源消费
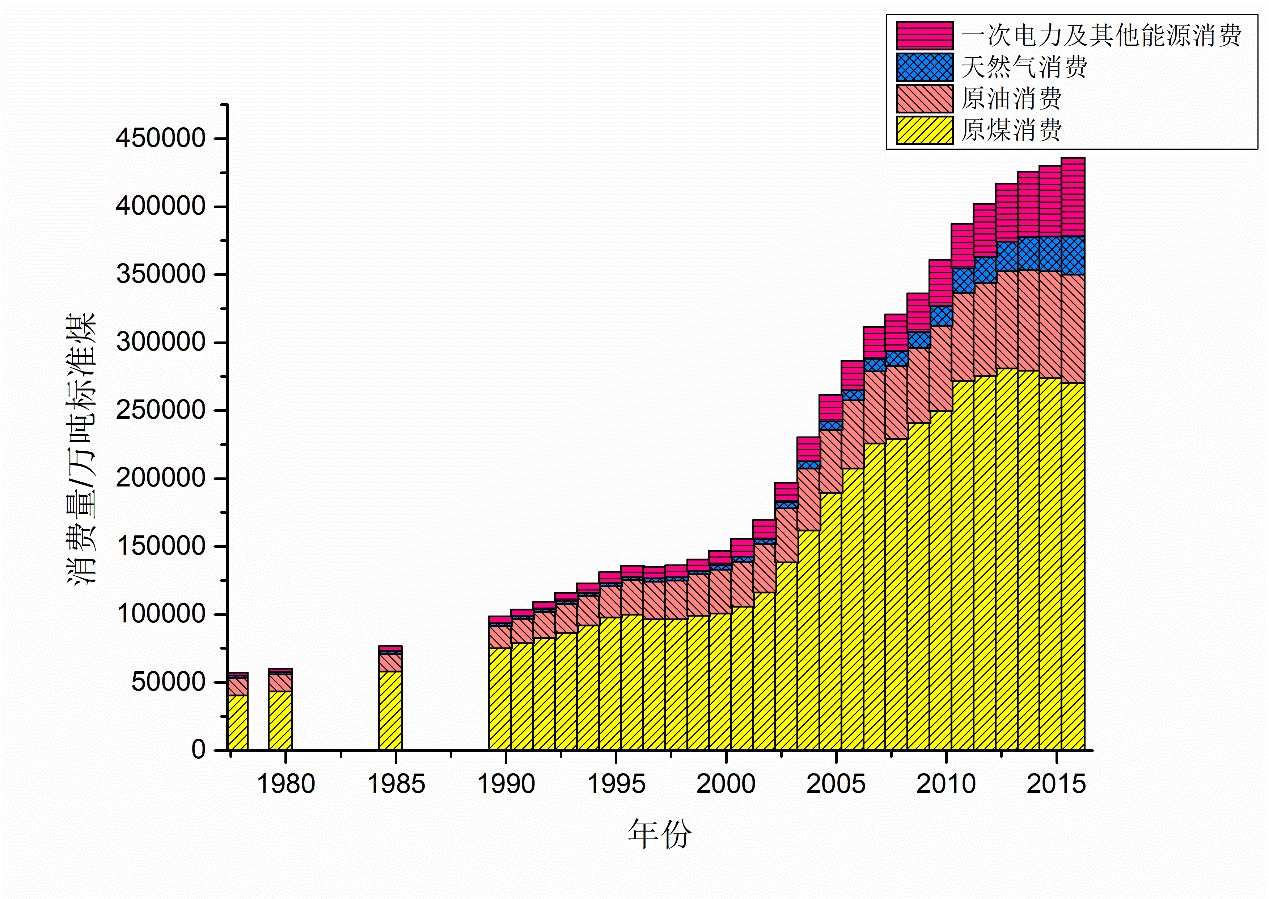
图 4.1.2 我国能源消费结构(截至2016年,数据来源:国家统计局《中国统计年鉴2017》)
2011年出版的《中国能源中长期(2030、2050)发展战略研究:综合卷》[^56]指出,我国要想保持长期的经济发展,必须解决相关领域的重大协调问题,特别是能源消费模式。该书给出了能源消费总量还将明显增加、需要用创新的观念和实践探索开拓新的能源消费模式、转变发展方式将十分急迫、我国基本国情决定了必须提高能效四个重要判断。
未来,我国的能源经济还将随着城镇化的进一步推进,人民生活水平的提高,在居民生活建筑用能、交通运输用能等方面呈现快速增长,同时我国高耗能工业的增速将不断放缓,水泥、钢铁和有色金属等都将在2020年前后达到峰值,纯碱、烧碱、乙烯等化工原材料将在2030年前出现峰值,同时鉴于节能技术和管理的发展,我国的工业能源消费有可能实现趋零增长(即保持总量稳定为一定值)。
小结
可以看出,我国的能源消费结构业已逐步开始变化,清洁能源比重不断上升,原油和煤炭的需求稳定,当然其在总的能源结构中占比减少。同时应当指出,我国的社会经济运行体制决定了我国的能源产业发展会更多地考虑到整个系统运行的稳定性控制,这不同于市场化程度高的国家更倾向于在新型能源方面的“开源”来解决高成本输电等能源供给中的问题,我国的能源产业发展更多的应是在已有的能源结构上,对于现有能源来源进一步优化和结构调整,适当引入新型能源,但着重提高现有能源的利用效率,即“碳效率”。这也是符合可持续发展要求和生态文明建设的。
而在我国能源战略[^58]中,为了深入推进能源革命、推动能源生产利用方式变革、优化能源供给结构、提高能源利用效率、建设清洁低碳安全高效的现代能源体系和维护国家安全,需要利用新技术和对已有技术的继续改善以推动能源结构优化升级,同时统筹能源网络建设、推进能源和信息领域新技术融合,并与交通通信等基础设施网络建设统筹发展并建成能源互联网。“十三五”规划在能源技术和基础研究方面,提到了智能电力系统、煤炭清洁高效利用、可再生能源(风电、光伏)、核电、非常规油气、能源输送通道、能源储备设施、能源关键技术装备等能源发展重大工程。下节从不同角度作简单介绍,并考虑理论计算的作用,略作分析,希望可以作为未来产业发展的参考。
煤炭利用技术
超临界水煤气化作为煤炭清洁利用的重要技术,其在我国的煤炭清洁利用中具有重要意义,而超临界水煤气化过程由于其可以解决包括环保、分离和联合发电等一系列问题而具有广泛的应用前景[^59],特别是西安交通大学动力工程多相流国家重点实验室主持的“煤炭超临界水气化制氢和H2O/CO2混合工质热力发电多联产基础研究”项目[^60],为这方面技术的应用转化提供了示范。
整体煤气化联合循环(integrated gasification combined cycle, IGCC)技术,是实现煤电厂碳减排和污染处理的一项重要应用,柳康等的研究[^61]依托于华能(天津)265MW级整体煤气化联合循环发电系统(IGCC)示范电站,针对IGCC技术中的低温耐硫变换系统、低温耐硫变换系统、PDS法硫回收系统进行了理论分析和系统工艺建模,并指出当产品为液态 CO2时,CO2捕集能耗为4.30GJ/t CO2,液态CO2成本约为341元/吨,当氢气增值为0.8元/m3(标况下)时,装置即可盈利。
IGCC和超临界技术是在我国煤炭清洁利用工业技术中重要的两个技术,其着眼于实际应用进行的技术研发和理论分析等,对于我国煤炭能源利用效率的提升具有重要意义。以上引述的实例中,并没有体现到基于传统意义上的计算化学的应用。但应当指出,我国当前的技术结构并不支持从微观到应用生产这种大规模的技术应用研究,但上述研究中的确存在有应用基本的计算化学研究成果的内容,如在其理论分析中就应用到了可由燃烧和多相反应热力学计算的平衡信息,进而建立完整的物料和能量平衡模型等。
尽管基于宏观的工艺生产处理和建模对于目前的市场和技术需求业已足够,不过未来如果要实现对于煤炭的更精细化利用、或是对于化工生产过程中某些关键催化技术或材料的改进研究仍需要通盘考虑从理论计算到工业级应用的联合。工业界在这方面的专利储备可能会逐渐出现。
可再生能源利用和新型电网技术
太阳能发电技术
高效晶硅太阳能电池发展的瓶颈在于其光衰的提高,光衰主要源于直拉单晶硅中少数载流子寿命的降低,在硼掺杂的直拉单晶硅中,这种少子寿命随时间的衰减现象更为明显,其与氧的一种亚稳缺陷密切相关,掺镓单晶硅技术是解决光衰并避免单晶利用不足的重要基础。[^62]在薄膜太阳能电池的研究中,严辉等[^63]通过银掺杂的铜锌锡硫薄膜太阳能电池吸收层实现了对于薄膜的瞬态光电流密度与稳态光电流密度的提高。太阳能电池硅片的等离子增强化学气相沉积法(PECVD)是太阳能电池片生产中的一种重要成膜方法,如东方日升新能源股份有限公司[^64]和应用材料公司(美)[^65]的相关专利技术分别用于SiN膜和P、B掺杂Si镀膜的生产应用。而以上应用技术研究中更多的是实验研究,可以发现其均有运用理论化学计算数据进一步细化研究,不断降低生产成本形成竞争力的潜力。
生物质液体燃料制取和利用技术
由水蒸气重整生物油制取的生物油合成气,用费托合成法直接合成洁净液体生物油燃料的研究[^66]指出,在300℃,1.5MPa的典型反应条件下可以实现36%的碳转化率和44%的C5+选择性,其研究中使用了MgO负载的C12A7-O-催化剂。而在生物油的转化中,浙江大学清洁能源利用国家重点实验室使用超临界技术、烘培预处理、相关锅炉研究开发和秸秆CFB项目建设进行的生物油制取高品位液体燃料技术和燃烧发电[^67]对于我国生物质资源的开发利用具有重要的示范意义,与此相关的环境影响亦有一定研究[^68]。上述实例中,涉及部分催化剂和前述章节中的燃烧反应基础理论,可以进一步与相关研究整合加强通用性的技术建设和系统化理论的形成。
面向未来的基础性技术
新型可再生能源转化概念和技术
本节简述我国研究人员关于微藻热解和天然气水合物开发二例。微藻与传统的纤维素生物质相区别的,其进行热解制备液体燃料甚至可以通过生物技术控制培养条件提高脂类含量以实现高的产率和高品质的热解油。[^69]而通过如水热、醇解、热解、氢化热解等不同反应条件的选择,也可以获得不同组成和产量的热解油。[^70]在天然气水合物开发中,我国基于能源安全的考虑,进行了可燃冰开采的技术尝试和攻关,勘探技术和测试分析技术业已比较成熟,但全世界范围内可燃冰开采尚不具备有商业竞争力[^71][^72]。考虑到这一领域的研究起步较晚,但却值得注意的是其开发研究中有用于开采模拟和风险控制的体系化计算模拟软件系统,尽管这种软件系统并非是理论化学软件,不过应当注意到当前商业应用技术研究对于模拟软件开发重要地位的判断。
基础科学
除去基于量子化学原理和化学信息学的统计分析技术理论,如要保障这些基于信息科学的技术高效运行和服务于经济社会发展,就需要发展超算、分子设计理论和应用程序和相应的信息处理计算理论的体系建设。尽管如此,需要指出,本文立足于化石能源、新能源等可用于提供能量,但并不抵触其作为化工原料的重要用途,而且事实上,将包括化石能源在内的能源物质用作化工原料更符合其利用价值,纯粹的能源应当是包括光伏、热电等产生的电能,本文更期待在未来可能出现技术突破与产业化的人工可控小型核聚变。
我国能源基础技术理论研究领域现状[^1]
我国能源产业的理论研究主要立足于由国家能源局设立的一系列国家能源研发(实验)中心,其涵盖了核电、风电、高效发电、输电、煤炭清洁转化与利用、能源勘探与开发、设备材料以及能源装备等能源重点行业和领域,也有跨学科、单位的协作大平台如大连洁净能源国家实验室、中科院广州能源研究所、中科院青岛生物能源与过程研究所、中科院北京纳米能源与系统研究所等。根据国家能源局2018年预算[^73],在科学技术支出中,即科技重大项目中的支出为70464.03万元,考虑到众多研究项目,这一数据其实并不算太多。而我国的能源化学教育方面和国际通行一样,均缺乏系统性的人才培养体系,一些文献认为没有能源化学专业化的人才培养是重要原因。而我国的能源化学技术转移和产业化更多的以技术供给方驱动为主,如专利权人的知识产权许可和技术合作开发,而不同于美国、欧洲和日本的委托研发和合作开发模式,一定程度上存在技术人员追求技术先进性而忽视技术产业化的问题。这一问题最近已有相关政策文件说明,但本文认为产生改善不仅需要时间,更需要产业界进一步的结构调整和配套数据信息服务的建设。能源化学专业的设立或许并不是解决相关问题的必要条件,培养和能源应用、研究信息服务及计算模拟相关的专业管理人员可能更有必要。
前景和展望
在“既要金山银山又要绿水青山”的环境友好型社会发展的需求下,我国的能源结构和能源化学也要充分考虑国际贸易中的碳减排压力和提高“碳生产率”,以提高我国在走向贸易大国过程中的国际竞争力。研究指出我国提高碳生产率的关键在于制造业的规模经济化、技术创新、行业升级、能源结构优化和差异化的推进不同行业的政策发展[^74]。
值得注意的是,在能源科学的技术研究中,大量企业在市场需求和技术进步的推动力下,进行理论研究的需求十分活跃,我国正处于经济结构调整和产业结构升级的关键时期,结合中国制造2025计划、创新驱动国家战略的背景,期望更多的政策和技术管理工作能注重于将高校和研究机构中的创新创造潜力,特别是理论研究力量整合起来,专注于某些特定方向的实际前景应用,打通从理论到实际技术发展的研究工作和应用有机循环,形成类似于以产业养研究的良性发展,保持这类研究成果转化中研究人员和应用技术转化人员的合理比例和良性互动,并依托于市场导向和政策前景及时推进技术改革,保障技术储备和促进经济转型,实现良好的社会效益。不仅限于高校科研人员的横向课题,更多的要是一些国民经济支柱产业企业和研究机构展开良好、无缝的合作,本文希望相关政策的发展能有利于这种合作模式,但考虑到经济运行规律,或许对企业来说,具有替代能力的新能源领域研究更引兴趣一些。
因此需要指出的是,要想实现能源科学领域的技术革新和产业升级,着实不是某个工程行业或高等教育行业通过鼓励技术研发就可以实现的,要实现整个国家的能源技术升级和可持续发展,要全面地鼓励科技创新、增加教育投入这类基础设施的建设,根本上要解决的是我国经济发展对于低成本人力资源、环境和能源资源仍存在的依赖,并转向技术和科技附加值增长,这就更加需要着力发展理论研究和提高基础学科水平,实现技术储备和研究人员、技术人员的持续发展。本文认为这需要决策者通盘考虑信息服务业对于制造业和重工业的作用,运用经济手段辅助政策手段的不断调节。
结论
本文浅述了和能源化学理论研究有关的关键理论、技术和应用进展,结合我国能源形势和研究、政策、产业发展现状,以及展望新的形势下我国能源科学和能源产业发展的趋势和动向,指出我国能源科学的持续健康发展需要整体政策设计的考量,要综合科技、教育、政策和基础设施建设等多方面的努力。结合能源化学理论研究的基础意义和我国能源技术的发展需求,指出要逐步引导技术储备的产生和人才力量的支持,发挥日趋重要的理论研究在产业发展中的重要作用,并适时整合相关企业和研究机构在市场需求和技术储备的发展,保障产业和研究的对接,促进研究和产业发展的良性互动。
最后需要指出的是,我国对于比较接近纯粹理论计算的需求看似尚不迫切。而其实这种基于基础理论的研究,特别是系统化的理论体系和生产应用对接机制的建立,在整个世界的科学体系中尚是初露矛头,仍需要相当长时间的研究攻关,在化学、物理学、信息科学、管理学甚至传播学、社会学的研究中都有其问题存在。不过本文确信,相关能源化学研究一旦有所突破,将极大地促进社会生产力的发展进步和相关的环保、服务产业和信息资源产业的综合发展,并令我国的综合国力有坚实的发展,对于保护国家安全也将是有力的帮助。
参考文献
[^1]: 国家自然科学基金委员会, 中国科学院. 中国学科发展战略·能源化学[M]. 北京: 科学出版社, 2018.
[^2]: 国家自然科学基金委员会, 中国科学院. 未来10年中国学科发展战略·化学[M]. 北京: 科学出版社, 2011.
[^3]: 王中平, 孙振平, 金明. 表面物理化学[M]. 上海: 同济大学出版社, 2015.
[^4]: LI D, NIELSEN M H, LEE J R, et al. Direction-specific interactions control crystal growth by oriented attachment[J]. Science, 2012, 336(6084): 1014-1018.
[^5]: TIAN N, ZHOU Z Y, SUN S G, et al. Synthesis of tetrahexahedral platinum nanocrystals with high-index facets and high electro-oxidation activity[J]. Science, 2007, 316(5825): 732-735.
[^6]: STAMENKOVIC V R, FOWLER B, MUN B S, et al. Improved oxygen reduction activity on Pt3Ni(111) via increased surface site availability[J]. Science, 2007, 315(5811): 493-497.
[^7]: RODRIGUEZ J A, MA S, LIU P, et al. Activity of CeOx and TiOx nanoparticles grown on Au(111) in the water-gas shift reaction[J]. Science, 2007, 318(5857): 1757-1760.
[^8]: HUO W T, LIN X, YU S, et al. Corrosion behavior and cytocompatibility of nano-grained AZ31 Mg alloy[J]. 2019, 54(5): 4409-4422.
[^9]: KIM J-J, CHOI Y, SURESH S, et al. Nanocrystallization During Nanoindentation of a Bulk Amorphous Metal Alloy at Room Temperature[J]. 2002, 295(5555): 654-657.
[^10]: 国家自然科学基金委员会, 中国科学院. 中国学科发展战略: 理论与计算化学[M]. 科学出版社, 2016.
[^11]: SCHWABE T, GRIMME S. Theoretical Thermodynamics for Large Molecules: Walking the Thin Line between Accuracy and Computational Cost[J]. Acc Chem Res, 2008, 41(4): 569-579.
[^12]: GOERIGK L, GRIMME S. A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions[J]. Physical Chemistry Chemical Physics, 2011, 13(14): 6670-6688.
[^13]: CRAMER C J, TRUHLAR D G. Implicit Solvation Models: Equilibria, Structure, Spectra, and Dynamics[J]. Chem Rev, 1999, 99(8): 2161-2200.
[^14]: STRUEBING H, GANASE Z, KARAMERTZANIS P G, et al. Computer-aided molecular design of solvents for accelerated reaction kinetics[J]. Nat Chem, 2013, 5: 952.
[^15]: BLIGAARD T, NøRSKOV J K, DAHL S, et al. The Brønsted–Evans–Polanyi relation and the volcano curve in heterogeneous catalysis[J]. Journal Of Catalysis, 2004, 224(1): 206-217.
[^16]: MICHAELIDES A, LIU Z P, ZHANG C J, et al. Identification of General Linear Relationships between Activation Energies and Enthalpy Changes for Dissociation Reactions at Surfaces[J]. J Am Chem Soc, 2003, 125(13): 3704-3705.
[^17]: JACOBSEN C J H, DAHL S, CLAUSEN B S, et al. Catalyst Design by Interpolation in the Periodic Table: Bimetallic Ammonia Synthesis Catalysts[J]. J Am Chem Soc, 2001, 123(34): 8404-8405.
[^18]: STUDT F, ABILD-PEDERSEN F, BLIGAARD T, et al. Identification of Non-Precious Metal Alloy Catalysts for Selective Hydrogenation of Acetylene[J]. 2008, 320(5881): 1320-1322.
[^19]: SKOPLYAK O, BARTEAU M A, CHEN J G. Reforming of Oxygenates for H2 Production: Correlating Reactivity of Ethylene Glycol and Ethanol on Pt(111) and Ni/Pt(111) with Surface d-Band Center[J]. The Journal of Physical Chemistry B, 2006, 110(4): 1686-1694.
[^20]: CIOBîCǍ I M, FRECHARD F, VAN SANTEN R A, et al. A DFT Study of Transition States for C−H Activation on the Ru(0001) Surface[J]. The Journal of Physical Chemistry B, 2000, 104(14): 3364-3369.
[^21]: LIU Z-P, HU P. General Rules for Predicting Where a Catalytic Reaction Should Occur on Metal Surfaces: A Density Functional Theory Study of C−H and C−O Bond Breaking/Making on Flat, Stepped, and Kinked Metal Surfaces[J]. J Am Chem Soc, 2003, 125(7): 1958-1967.
[^22]: VAN SANTEN R A. Complementary Structure Sensitive and Insensitive Catalytic Relationships[J]. Acc Chem Res, 2009, 42(1): 57-66.
[^23]: SHETTY S, JANSEN A P J, VAN SANTEN R A. CO Dissociation on the Ru(112̅1) Surface[J]. The Journal of Physical Chemistry C, 2008, 112(36): 14027-14033.
[^24]: MAVRIKAKIS M, BäUMER M, FREUND H-J, et al. Structure Sensitivity of CO Dissociation on Rh Surfaces[J]. 2002, 81(3): 153-156.
[^25]: ANISIMOV V I, ZAANEN J, ANDERSEN O K. Band theory and Mott insulators: Hubbard U instead of Stoner I[J]. Physical Review B, 1991, 44(3): 943-954.
[^26]: ARYASETIAWAN F, GUNNARSSON O. The GW method[J]. Reports on Progress in Physics, 1998, 61(3): 237.
[^27]: SCHULZ H. Short history and present trends of Fischer–Tropsch synthesis[J]. Applied Catalysis A: General, 1999, 186(1): 3-12.
[^28]: GEERLINGS J J C, WILSON J H, KRAMER G J, et al. Fischer–Tropsch technology — from active site to commercial process[J]. Applied Catalysis A: General, 1999, 186(1): 27-40.
[^29]: BRADY R C, PETTIT R. Mechanism of the Fischer-Tropsch reaction. The chain propagation step[J]. J Am Chem Soc, 1981, 103(5): 1287-1289.
[^30]: BILOEN P, HELLE J N, VAN DEN BERG F G A, et al. On the activity of Fischer-Tropsch and methanation catalysts: A study utilizing isotopic transients[J]. Journal Of Catalysis, 1983, 81(2): 450-463.
[^31]: KUMMER J, PODGURSKI H, SPENCER W, et al. Mechanism Studies of the Fischer—Tropsch Synthesis. The Addition of Radioactive Alcohol[J]. 1951, 73(2): 564-569.
[^32]: PICHLER V H, SCHULZ H J C I T. Neuere Erkenntnisse auf dem Gebiet der Synthese von Kohlenwasserstoffen aus CO und H2[J]. 1970, 42(18): 1162-1174.
[^33]: CAO D-B, LI Y-W, WANG J, et al. Chain growth mechanism of Fischer–Tropsch synthesis on Fe5C2(001)[J]. Journal of Molecular Catalysis A: Chemical, 2011, 346(1): 55-69.
[^34]: RIEDMüLLER B, CIOBı̂CĂ I M, PAPAGEORGOPOULOS D C, et al. CO adsorption on hydrogen saturated Ru(0001)[J]. 2001, 115(11): 5244-5251.
[^35]: LOVELESS B T, BUDA C, NEUROCK M, et al. CO Chemisorption and Dissociation at High Coverages during CO Hydrogenation on Ru Catalysts[J]. J Am Chem Soc, 2013, 135(16): 6107-6121.
[^36]: CIOBICA I M, KLEYN A W, VAN SANTEN R A. Adsorption and Coadsorption of CO and H on Ruthenium Surfaces[J]. The Journal of Physical Chemistry B, 2003, 107(1): 164-172.
[^37]: CHEN Y-H, CAO D-B, JUN Y, et al. Density functional theory study of CO adsorption on the Fe (1 1 1) surface[J]. 2004, 400(1-3): 35-41.
[^38]: RUSCIC B, PINZON R E, VON LASZEWSKI G, et al. Active Thermochemical Tables: thermochemistry for the 21st century[C]// Active Thermochemical Tables: thermochemistry for the 21st century. Journal of Physics: Conference Series. IOP Publishing,16: 561.
[^39]: GOLDSMITH C F, MAGOON G R, GREEN W H J T J O P C A. Database of small molecule thermochemistry for combustion[J]. 2012, 116(36): 9033-9057.
[^40]: CôME G, WARTH V, GLAUDE P, et al. Computer-aided design of gas-phase oxidation mechanisms—application to the modeling of n-heptane and iso-octane oxidation[C]// Computer-aided design of gas-phase oxidation mechanisms—application to the modeling of n-heptane and iso-octane oxidation. Symposium (International) on Combustion. Elsevier,26: 755-762.
[^41]: VAN DUIN A C, DASGUPTA S, LORANT F, et al. ReaxFF: a reactive force field for hydrocarbons[J]. 2001, 105(41): 9396-9409.
[^42]: CHENOWETH K, VAN DUIN A C, GODDARD W A J T J O P C A. ReaxFF reactive force field for molecular dynamics simulations of hydrocarbon oxidation[J]. 2008, 112(5): 1040-1053.
[^43]: GONG J, LUQUE R. Catalysis for production of renewable energy[J]. Chem Soc Rev, 2014, 43(22): 7466-7468.
[^44]: ZHANG Z C, XU B, WANG X. Engineering nanointerfaces for nanocatalysis[J]. Chem Soc Rev, 2014, 43(22): 7870-7886.
[^45]: JELOAICA L, SIDIS V. DFT investigation of the adsorption of atomic hydrogen on a cluster-model graphite surface[J]. Chemical Physics Letters, 1999, 300(1): 157-162.
[^46]: PISANI C, CAUSà M, DOVESI R, et al. Hartree-fock ab-initio characterization of ionic crystal surfaces with a slab model. The (0001) face of α-Al2O3[J]. Progress in Surface Science, 1987, 25(1): 119-137.
[^47]: CAO Y, CHEN Z-X. Slab model studies of water adsorption and decomposition on clean and X- (X = C, N and O) contaminated Pd(111) surfaces[J]. Physical Chemistry Chemical Physics, 2007, 9(6): 739-746.
[^48]: MARRINK S J, DE VRIES A H, MARK A E. Coarse Grained Model for Semiquantitative Lipid Simulations[J]. The Journal of Physical Chemistry B, 2004, 108(2): 750-760.
[^49]: REITH D, PüTZ M, MüLLER-PLATHE F. Deriving effective mesoscale potentials from atomistic simulations[J]. 2003, 24(13): 1624-1636.
[^50]: IZVEKOV S, VIOLI A, VOTH G A. Systematic Coarse-Graining of Nanoparticle Interactions in Molecular Dynamics Simulation[J]. The Journal of Physical Chemistry B, 2005, 109(36): 17019-17024.
[^51]: 侯怀宇, 张新平. 材料科学与工程中的计算机应用[M]. 国防工业出版社: 北京, 2015.
[^52]: 李莉, 王香. 计算材料学[M]. 哈尔滨: 哈尔滨工业大学出版社, 2017.
[^53]: 国家自然科学基金委员会, 中国科学院. 未来10年中国学科发展战略·材料科学[M]. 北京: 科学出版社, 2012.
[^54]: ONG S P, RICHARDS W D, JAIN A, et al. Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis[J]. Computational Materials Science, 2013, 68: 314-319.
[^55]: 王安建, 王高尚, 陈其慎, 等. 能源与国家经济发展[M]. 北京: 地质出版社, 2008.
[^56]: 中国能源中长期发展战略研究组. 中国能源中长期(2030、2050)发展战略研究:综合卷[M]. 科学出版社, 2011.
[^57]: 崔民选, 王军生, 陈义和. 中国能源发展报告.2015[M]. 北京: 社会科学文献出版社, 2015.
[^58]: 全国人民代表大会. 中华人民共和国国民经济和社会发展第十三个五年规划纲要[EB/OL] [2018.12.08]. http://www.xinhuanet.com//politics/2016lh/2016-03/17/c_1118366322.htm.
[^59]: 陈哲文. 超临界水煤气化方法及发电系统集成[D]. 中国科学院大学(中国科学院工程热物理研究所), 2018.
[^60]: 郭烈锦, 赵亮, 吕友军, 等. 煤炭超临界水气化制氢发电多联产技术[J]. 工程热物理学报, 2017, 38(03): 678-679.
[^61]: 柳康, 许世森, 李广宇, 等. 基于整体煤气化联合循环的燃烧前CO_2捕集工艺及系统分析[J]. 化工进展, 2018, 37(12): 4897-4907.
[^62]: 杨淑云, 李宁, 丰云恺, 等. 高效晶硅太阳电池的发展与抑制光衰的研究进展 [J]. 硅酸盐通报, 2013, 32(12): 2533-2537.
[^63]: 李桐, 张林睿, 杨炎翰, 等. 铜锌锡硫薄膜太阳能电池吸收层的银掺杂 [J]. 材料工程, 2018, (12): 95-100.
[^64]: 葛竖坚, 翟贝贝, 魏晓波, 等. 太阳能电池硅片的等离子增强化学气相沉积法, CN106282965A [P/OL]. 2017-01-04].
[^65]: S·盛, L·张, Z·袁, 等. 制造硅异质结太阳能电池的方法与设备, CN107142460A [P/OL]. 2017-09-08].
[^66]: 袁丽霞, 王兆祥, 董婷, 等. 生物质制取氢气和生物油液体燃料(英文) [J]. 中国科学技术大学学报, 2008, (06): 668-673.
[^67]: 骆仲泱. 生物质制取高品位液体燃料及燃烧发电[C]// 生物质制取高品位液体燃料及燃烧发电. 2014中国(国际)生物质能源与生物质利用高峰论坛(BBS 2014), 勤哲文化传播(上海)有限公司, 中国上海. 30.
[^68]: 瞿婷婷, 肖军, 沈来宏. 生物质制取高品位液体燃料的生命周期评价[J]. 太阳能学报, 2014, 35(09): 1700-1707.
[^69]: 常春, 孙培勤, 孙绍晖, 等. 我国生物质能源现代化应用前景展望(二)——生物质制备液体燃料的转化途径[J]. 中外能源, 2014, 19(07): 16-24.
[^70]: 王兵, 段培高. 微藻热化学转化制备液体燃料[C]// 微藻热化学转化制备液体燃料. 河南省化学会2014年学术年会, 河南省化学会, 中国河南郑州. 1.
[^71]: 唐志远, 胡云亭, 郭清正, 等. 天然气水合物勘探开发新技术进展 [J]. 地球物理学进展, 2015, 30(02): 805-816.
[^72]: 李丽松, 苗琦. 天然气水合物勘探开发技术发展综述 [J]. 天然气与石油, 2014, 32(01): 67-71+11-12.
[^73]: 国家能源局. 国家能源局部门预算[EB/OL] [2018-04]. http://zfxxgk.nea.gov.cn/auto80/201804/P020180413544574953616.pdf.
[^74]: 李小平, 杨翔, 谢振, et al. 低碳约束下的国际贸易发展新论[M]. 北京: 中国环境出版社, 2017.